
Help
Summary
1.
What is Pred-O3?
Pred-O3 is a web server that allows to explore interactions between molecules-odors and molecules-olfactory receptors. The server integrates a large database of odorant molecules tested on human, mouse and rat olfactory receptors. Furthermore, it allows to predict molecule-odor and molecule-olfactory receptor interactions.
Several tools have been previously developed and integrated into the server in order to deepen the knowledge on the mechanisms that link these interactions, including the possibility to search for compounds according to their structure, to predict the odorant potential of a molecule or olfactory receptors in interaction or to apply a docking protocol.
2.
Overview
On Pred-O3 you can access the annotations of the database with descriptive pages :
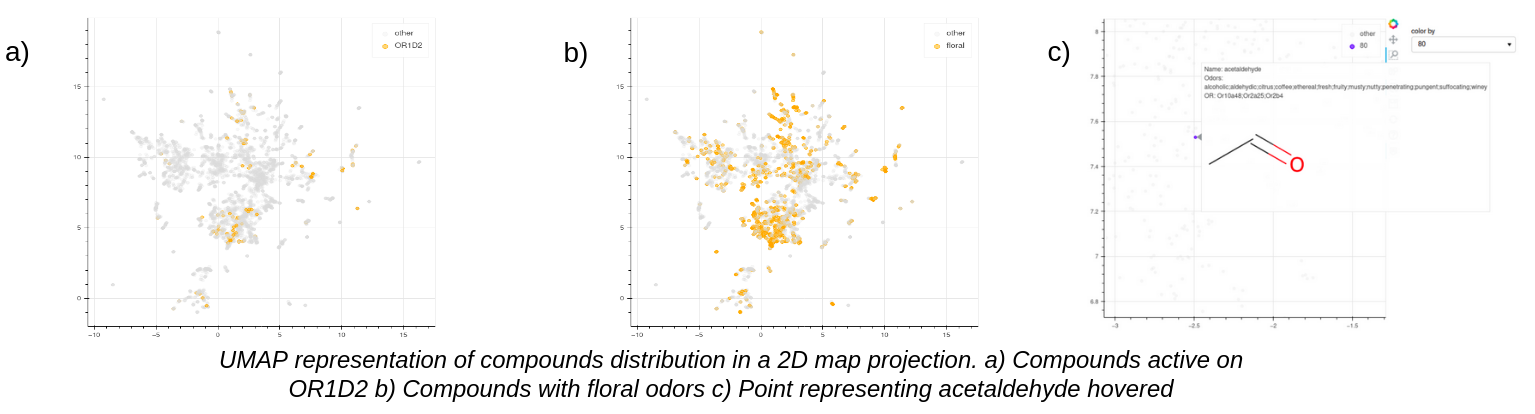
- Chemical descriptive page : Access to annotations for a given chemical. The user can find common chemical information, odor and olfactory receptor annotations as well as the phylogenic tree highlighting the interacting olfactory receptors, the chemical space.
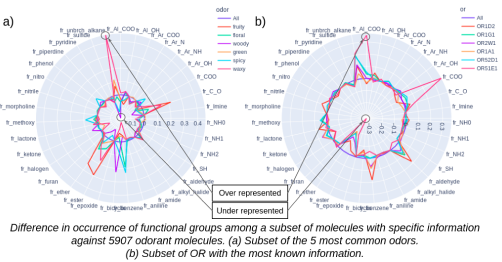
- Olfactory receptor descriptive page: Access annotations for a given olfactory receptor. The user can find common information on the protein, annotations on chemicals interacting with the olfactory receptor as well as the phylogenic tree highlighting olfactory receptors, a radar plot highlighting the under/overrepresented functional groups of the interacting chemicals.
- Odor descriptive page: Access the annotations for a given odor note. The user can find the chemicals known to have the given odor as well as the over/undererepresented functional groups of this list of chemicals and the chemical space.
The webserver includes 3 tools:
- Structure search : A tool for searching chemical of the database according to a chemical structure. The search is done according to 2 protocols. A protocol by similarity according to tanimoto score and a protocol according to the search of sub structures.
- Prediction : A tool to predict the potential odors or olfactory receptors for a molecule.
- Docking : A tool to link our database of olfactory receptors and odorant molecules to the SeamDock server in order to launch a docking protocol that predict the binding mode of a molecule in a selected olfactory receptor.
With these tools, graphical interfaces are implemented to support and deepen knowledge of odorant, olfactory receptors and odors:
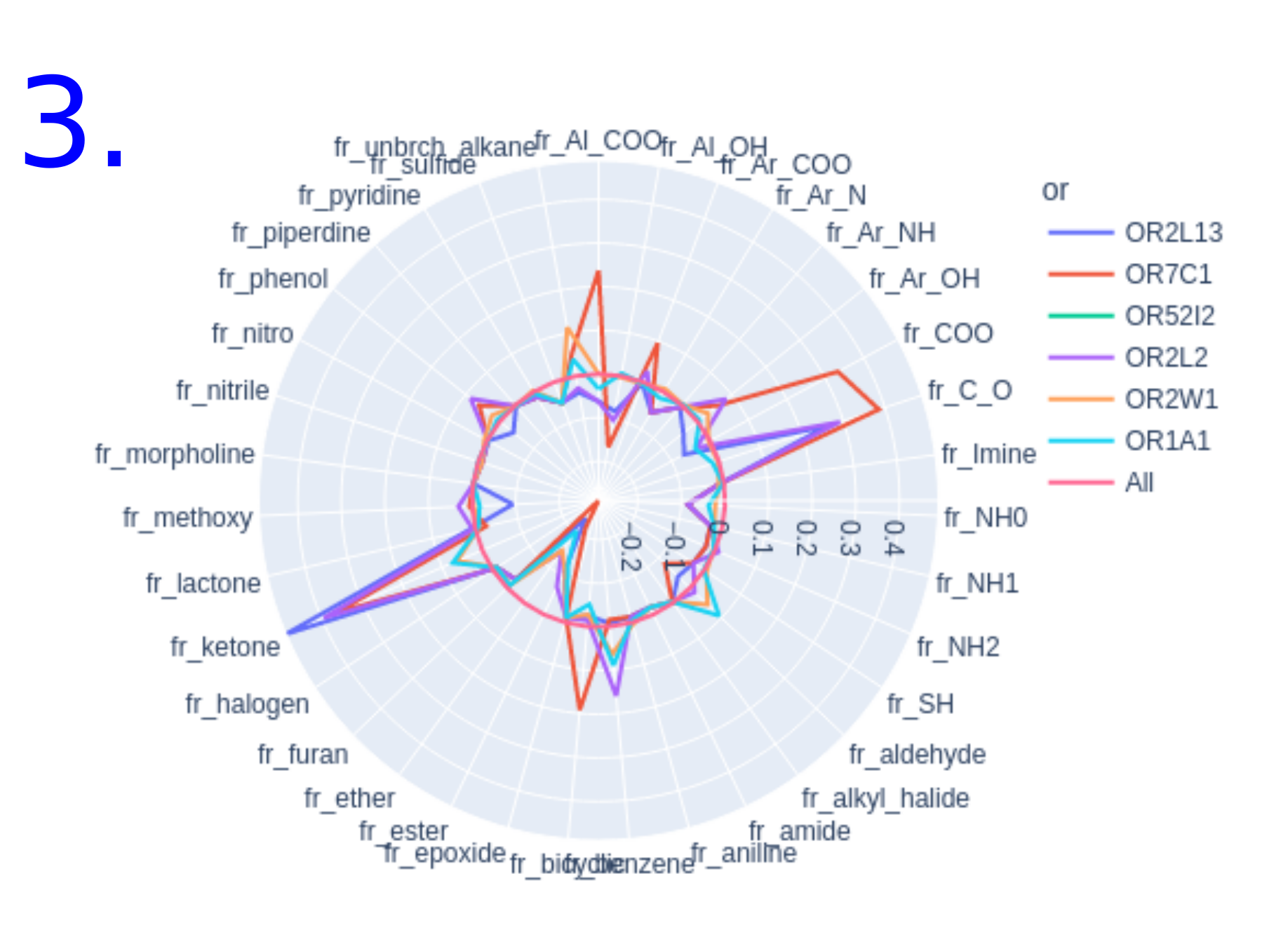
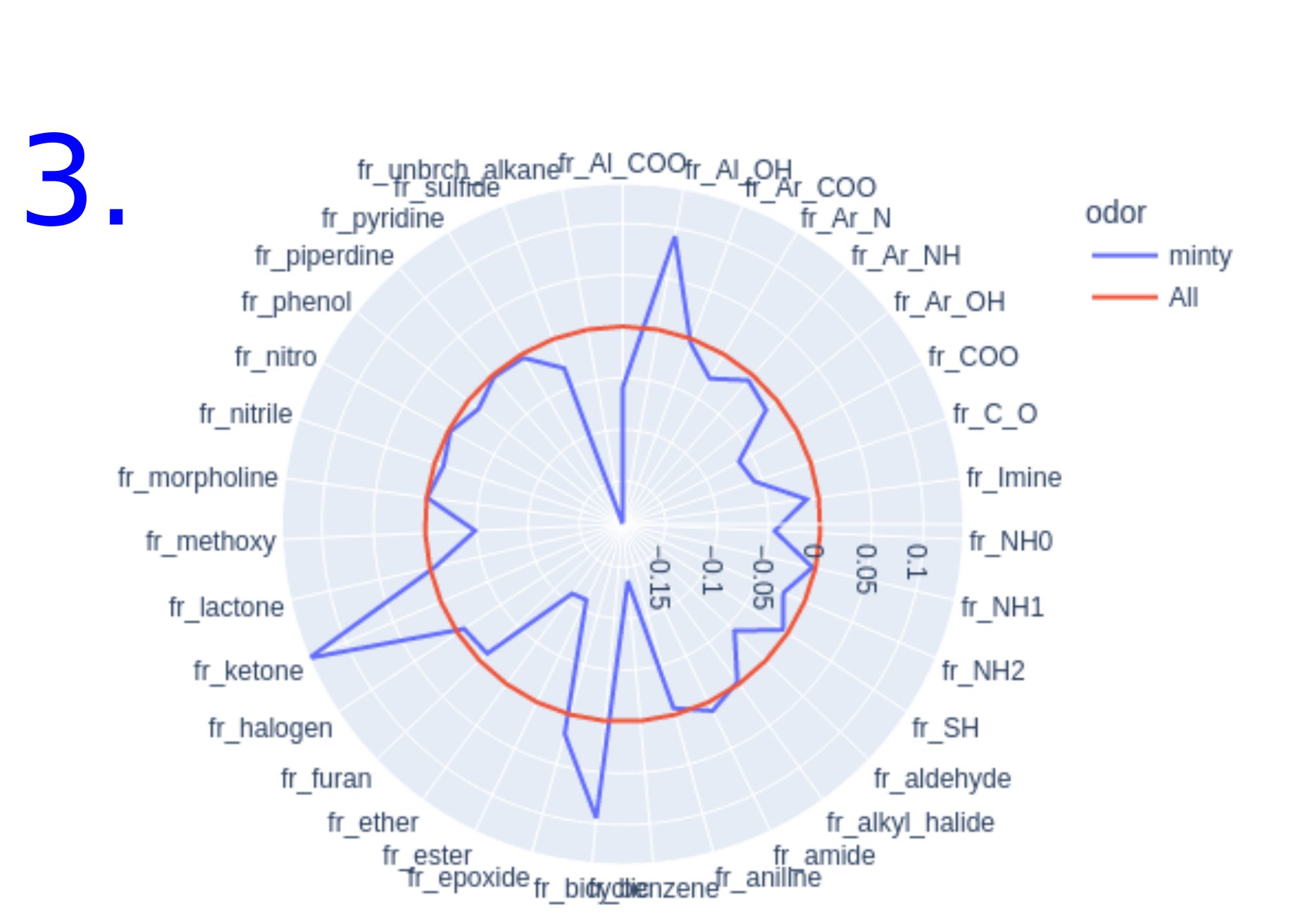
- Overrepresented/underrepresented functional groups : Radar plot of functional group occurrence. The occurrence of functional groups is calculated on the data set of 5908 molecules (odorant molecules and molecules with know interactiong olfactory receptors without known odors) and compared to a subset of molecules having an interesting odor or interacting with a specific olfactory receptor. The line of occurrence calculated over all molecules in the database is used as a reference and marked "All". Any line above the reference "All" represents over-represented functional groups and any line below the reference "All" represents an under-represented functional group. By hovering over the screen, additional information about the occurrence of the functional group in the subset of molecules is displayed.
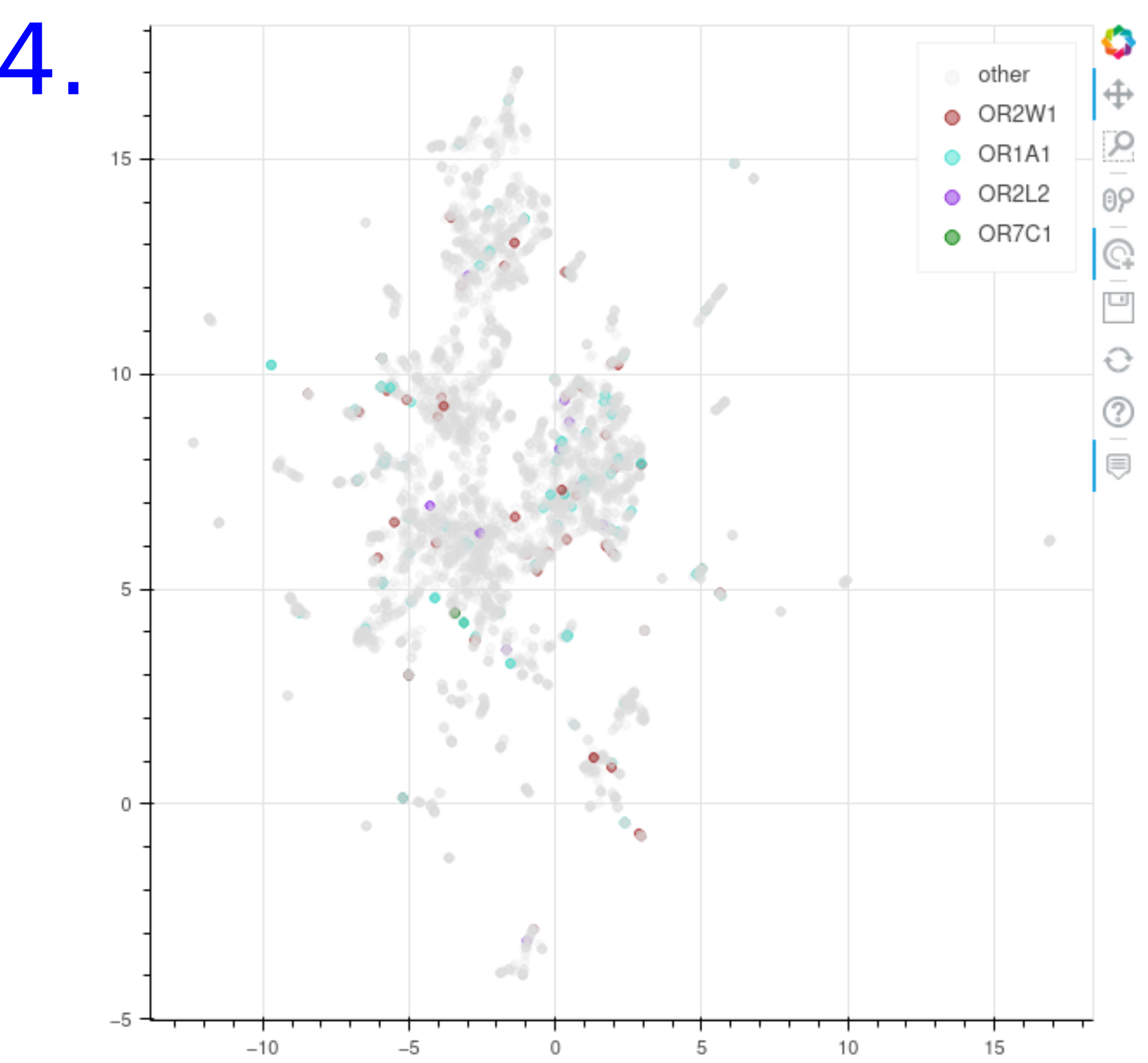
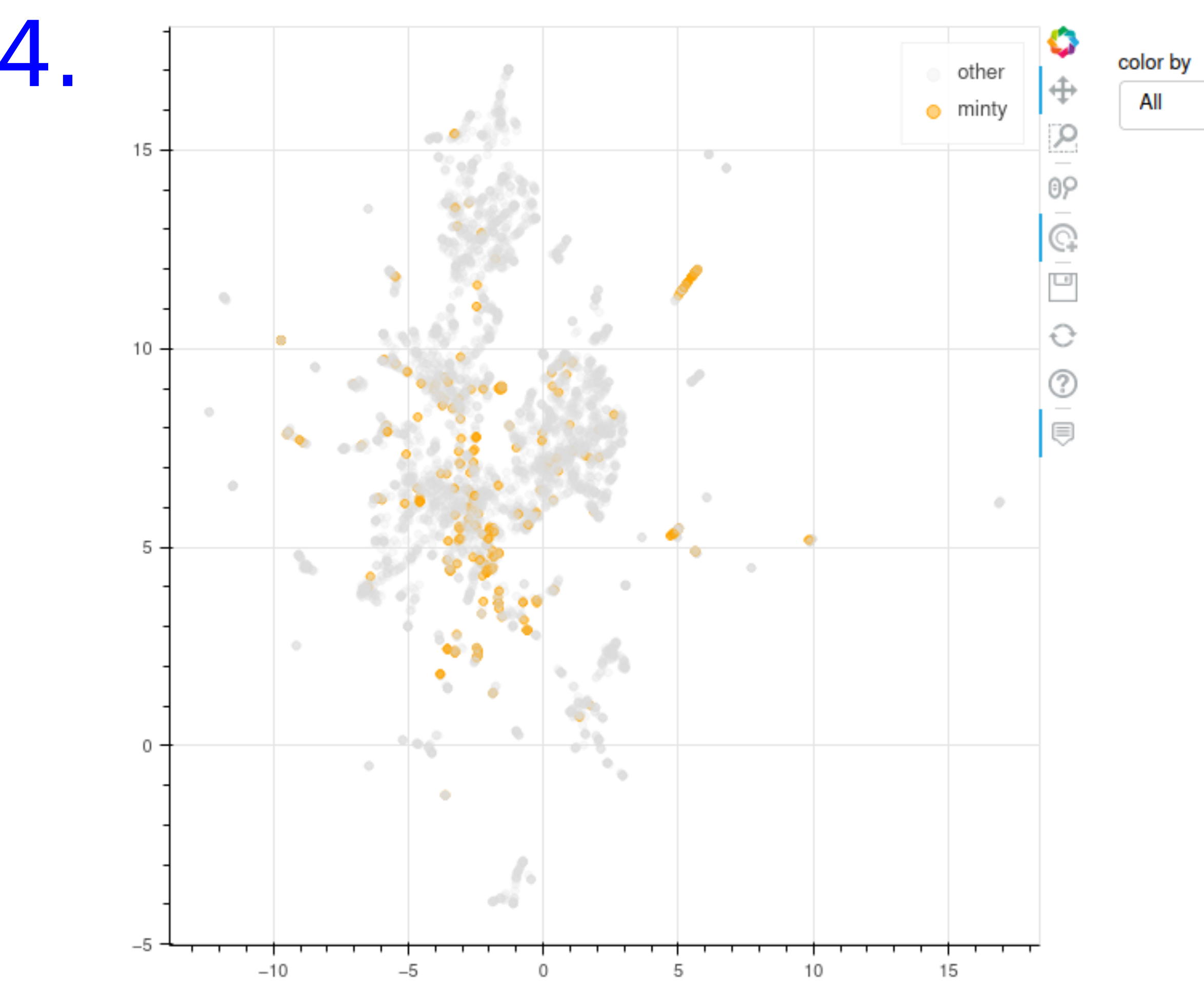
- Chemical Space : The similarity between sub groups of molecules sharing the same odors or interacting with the same olfactory receptors can be visualized through an UMAP representation in a 2d map projection. The molecules were carachetrised using the morgan fingerprint. When a point is hovered more information on the molecule and its odors and its olfactory receptors are displayed and the user can click on the dot to be redirected to the corresponding compound
- Phylogenetic tree : Mouse phylogenic tree was computed using the sequence of mouse olfactory receptors from HORDE website. Human phylogenic tree was computed using the sequence of human olfactory receptors from UniProt. The phylogenic tree was computed on the ngphylogeny website using the PhyML/OneClick workflow. Olfactory receptors were colored according their familly described by Glusman et al. The olfactory receptors in relation to a chemical are displayed bigger.
3.
Search
The user can search chemical, olfactory receptor and odor annotations directly on the home page or on the Search page.
The search panel displays several options:
- Search chemical annotations on the website
- Search olfactory receptor annotations on the website
- Search odor annotations on the website
- Examples to try the search protocol
- Launch the search protocol
The search protocol returns a table of chemicals, olfactory receptors or odors according to the selected query. The table highlights the query pattern in bold.
For chemical search, the query pattern is compared to name, case number, PubChem cid, smile, InChi and InChi key.
For olfactory receptor search, the query pattern is compared to the gene symbol, the UniProt id, synonyms and species.
For the odor search, the query pattern is compared to odor notes.
When searching for a chemical, an olfactory receptor or an odor in the home page, suggestions are available and clickable this will redirect the user to the descriptive page of interest.



4.
Compound-odor and compound-receptor annotations
Molecule
The Chemical descriptive page consists of different information:
- The 2D image structure of the chemical computed with RDKit
- General information of the chemical retrieved from PubChem: Name, synonyms, IUPAC name, CAS N°, molecular formula, molecular weight, smiles, InChi and InChi key
- The 3D structure of the molecule computed using RDKIt with the ETDKG (v3) force field
- List of odor notes of the molecule if any
- List of interacting olfactory receptors if any
- The chemical space with the molecule colored
- The pylogenetic tree of the interacting olfactory receptors highlighted if any
Olfactory receptor
The Olfactory receptor descriptive page consist of different information:
- General information on the olfactory receptor with its symbol (following Glusman et al. nomenclature ), UniProt ID, synonyms, species
- The 3D structure of the molecule computed and retrieved from AlphaFold (dec 2022 v4)
- List of chemicals known to interact with the olfactory receptor
- Radar plot of the overrepresented/ underrepresented functional groups among the chemicals interacting with the olfactory receptor
- The chemical space with the molecules interacting with the olfactory receptor colored
- The pylogenetic tree with the olfactory receptor highlighted
Odor
The Odor descriptive page is composed of different information:
- List of chemicals known to to have this odor note
- Radar plot of the overrepresented/ underrepresented functional groups among the chemicals having this odor
- The chemical space with the molecules having this odor highlighted
5.
Tools:
5.1.
Structure search
The server allows to search the database for molecular structures of interest in the form of a molecular similarity search and a sub structure search.
The similarity search allows to calculate and identify the structural similarity between the molecules of the database and a molecule of interest. The similarity is calculated according to a Tanimoto score based on the MACC fingerprints. MACC fingerprint is a bit vector of 120 bit counting the presence (1) or absence (0) of a specific functional group in the structure of the molecule. The tanimoto score varies between 0 and 1 the higher the score the higher the similarity between the molecules. Molecules are then sorted from most similar to the lowest.
The substructure search allows to search for a substructure pattern matching by searching via a smart pattern or any molecule identifier available in the searching method. It returns all the molecules of the database with the query substructure in its structure.
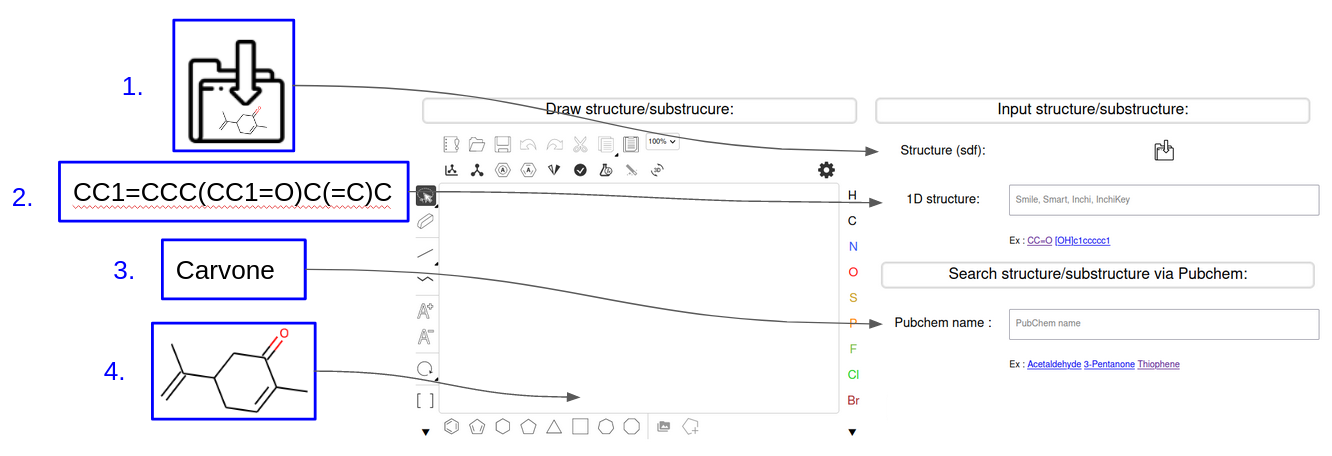
INPUT
To do a structural search by calculating the similarity the user must provide the structure of a molecule. Several possibilities are proposed to him to provide this molecule:
- The user can provide his structure as an .sdf file
- The user can provide the chemical structure under the form of a smile, InChi or InChi key
- The user can search for a molecule via its name which will be identified via a request sent to the PubChem server
- Sketcher : The user can draw his molecule of interest using the dedicated sketcher
CHOOSE PROTOCOL
Choose the similarity protocol or the substructure protocol by clicking on the launch button of your choice:

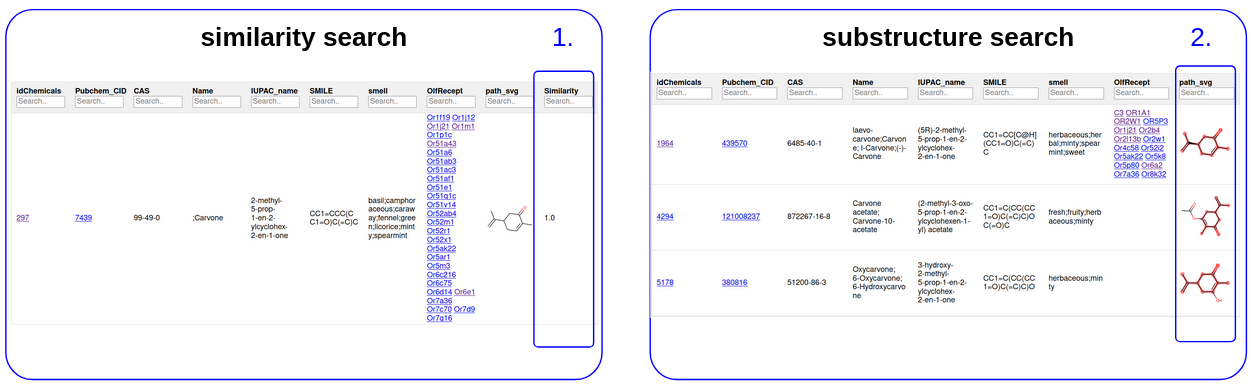
OUTPUT
The output results consist of a summary of the query compound if retrieved on PubChem server or the query pattern if not and a table of the molecules.

The results table shows the information on the molecules as well as their known odor and their interactive olfactory receptor if the information is available. In relation to the search protocol, two columns are possible:
- The tanimoto score for all the molecules of the database
- The molecules containing the substructure with the substructure highlighted in red
5.2.
Prediction
Two deep learning prediction models based on a graphical Graphical Neural Network (GNN) developed by Achebouche et al were added to the website to predict the potential odor and/or potential interacting human olfactory receptors of a query compound. The odor predictor can predict one or more odor classes among a set of 23 odor notes with a prc auc of 0.67 and the human olfactory receptor predictor can predict the potential interaction of a molecule with one or multiple human olfactory receptors among a set of 74 human OR with a prc-auc of 0.91.
The similarity of the query compound is also computed on all odorant of the database and the 10 most similar odorant are displayed with their odor notes and potential olfactory receptors interaction informations to strengthen the confidence of the given prediction.
INPUT
To perform a prediction the user must provide the structure of a molecule. Several possibilities are proposed to him to provide this molecule:
- The user can provide it’s structure as a .sdf file
- The user can provide the chemical structure under the form of a smile, InChi or InChi key
- The user can search for a molecule via its name which will be identified via a request sent to the PubChem server
- Sketcher : The user can draw his molecule of interest using the dedicated sketcher
CHOOSE MODEL
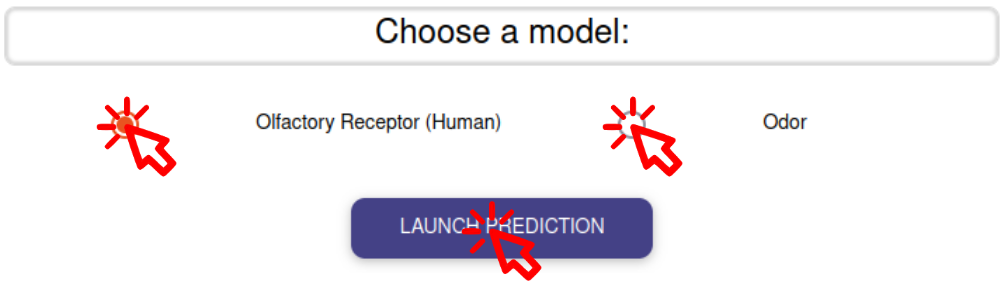
Select the model to predict the odor or the potential interacting olfactory receptors then launch the prediction by clicking on the blue button.
OUTPUT
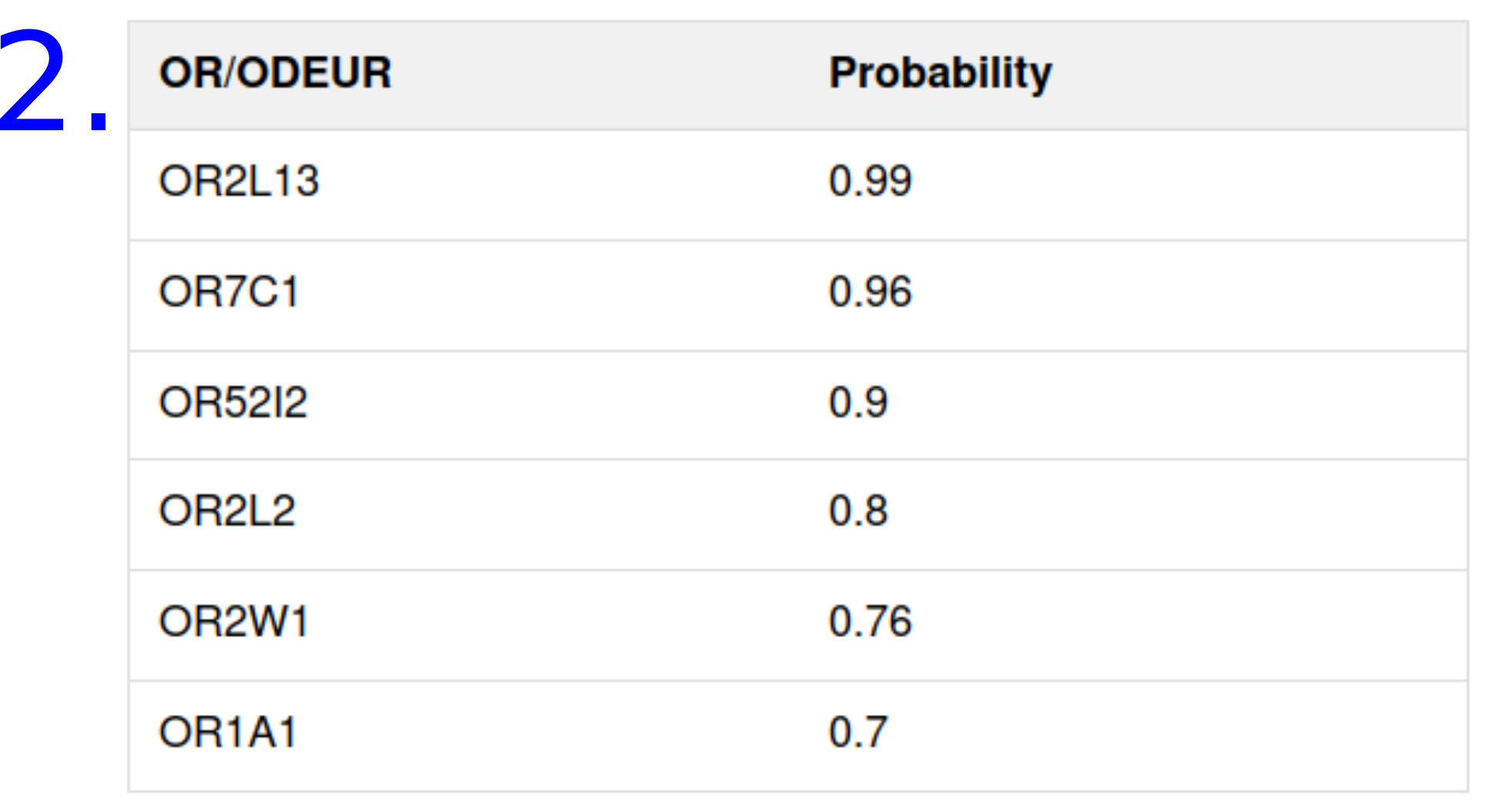
The output consist of several tables and figures:
- Physico-chemical information on the query molecule with it's molecular weight, the number h-bonds acceptors and h-bond donors atoms, the number of heavy atoms (non hydrogens), the number of rotatable bonds, the Topological Surface Area and the LogP.
-
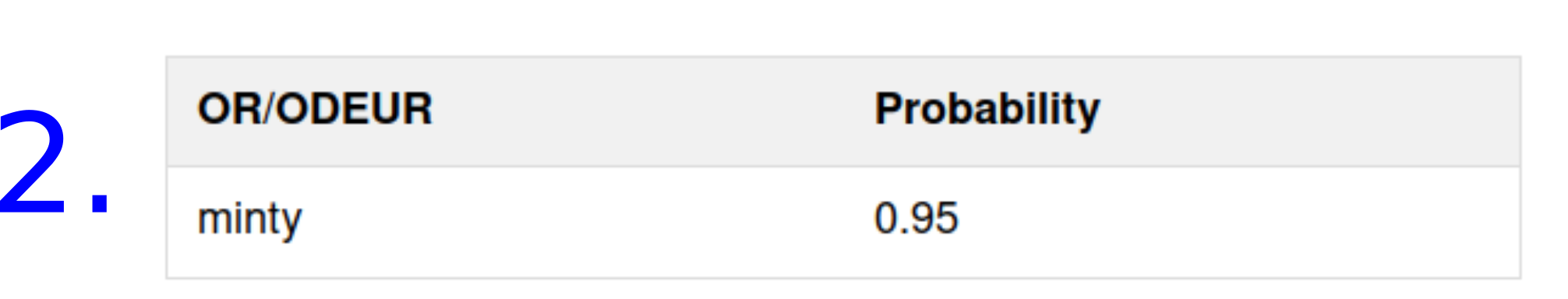
The predicted probabilities
- For the predicted odors
- For the predicted human olfactory receptors
-
Radar plot of the functional groups over/under represented among
- The set of molecules known to have the predicted odor
- The set of molecules known to interact with the predicted olfactory receptors
-
UMAP of the chemical space with chemicals colored by:
- The set of molecules known to have the predicted odor
- The set of molecules known to interact with the predicted olfactory receptors
- Table of the 10 most similar chemicals of the database with their known odors and their interacting olfactory receptors if any known
PREDICTION OUTPUT EXAMPLE FOR CARVONE:


5.3.
Docking
We linked the web server to the SeamDock webserver which allows us to perform online docking.
To perform docking we retrieved 3D structure of olfactory receptors computed with Alpha Fold for the proteins of the rat, mouse and human.
In order to standardize docking parameters for all docking of the ORs that are GPCR protein, we aligned all the structures against Or10a4 (UniProt id Q7TRM9) on the Z axis with a reference center set at 0.0.0 with pymol using TM-align. The grid box size has been set to X: 43 Å, Y: 24 Å, Z: 38 Å and centered in 0;0;0 in order to be sufficiently large and restricted to target the core of all the olfactory receptors.
seamdock is an online service that integrates multiple docking tools in a common framework allowing to easily perform ligand protein docking without advanced computer and docking knowledge.
INPUT
To perform the docking the user must choose the chemical structure to dock and the olfactory receptor to be docked on. Several possibilities are proposed to provide these structures:
- The user can provide it’s chemical structure as an .sdf file
- The user can provide the chemical structure under the form of a smile, InChi or InChi key
- The user can search for a molecule via its name which will be identified via a request sent to the PubChem server
- The user can retrieve a chemical from the database by its name, cas, synonyms
- The user can retrieve an olfactory receptor by it’s name or UniProt id
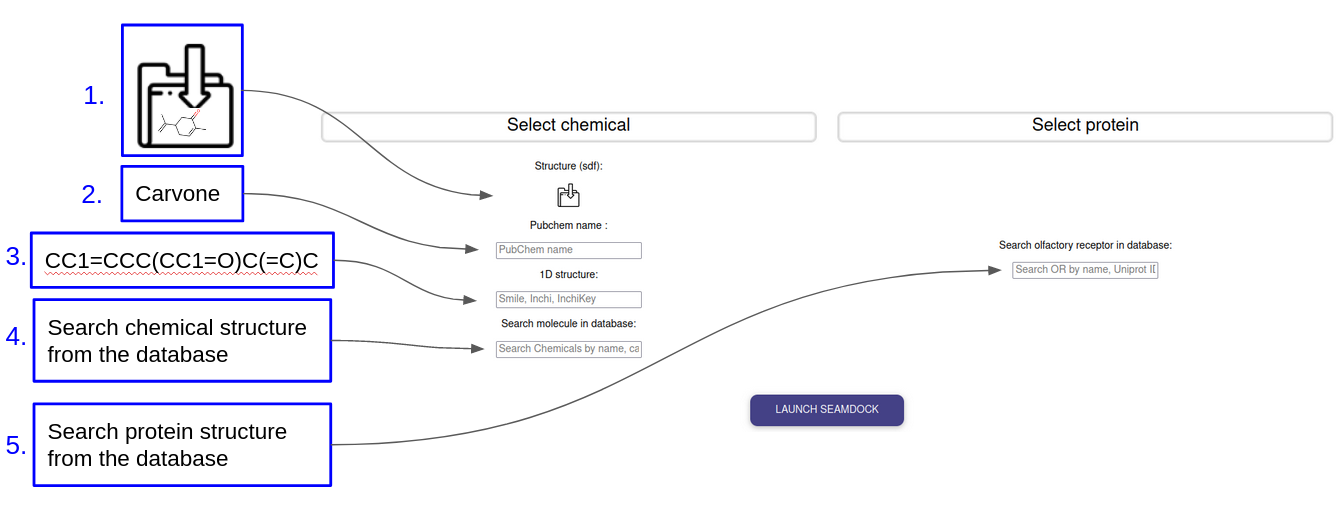
The user is redirect on the SeamDock server with parameters of the docking preregistered
6.
FAQ
Q1. Can we trust the docking results ?
A1. Chemicals and proteins structures were retrieved computationally, to date there is no 3D structure of olfactory receptors on the pdb so the results of docking method should be taken with caution.
7.
Browser compatibility
| OS | Version | Chrome | Firefox | Microsoft Edge | Safari |
| Linux | Ubuntu 20 | 87.0 | 107.0 | n/a | n/a |
| MacOS | Mojave | 87.0 | 84.0 | n/a | 16.6.1 |
| Windows | 10 | 87.0 | 84.0 | 116 | n/a |